Molecular Design: The Archetypical Inverse Design Challenge
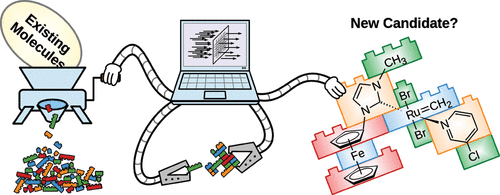
We envision a future in which molecular and materials discovery is realized via “Big Computing / Big Data”, not serendipity. We pursue this vision by developing fundamental and truly general-purpose methods for automated de novo design of any kind of molecule, including catalysts and other metal-containing functional molecules.
Discovery of functional molecules such as drugs and catalysts represents the archetypical inverse design challenge: Instead of starting with a specific molecular structure and then determining its properties, inverse design begins with the desired properties and tries to find structures that best meet those criteria. This is an immense challenge. Due to the numerous ways in which atoms of different elements such as carbon, hydrogen, and oxygen can be assembled into molecules, the chemical space is unfathomably large. Even the restricted chemical space of small, drug-like compounds is estimated to contain between 1023 and 1063 molecules.
Computational or experimental screening of any significant fraction of these molecules is impossible. Instead, AI-based optimization methods may circumvent the combinatorial explosion and navigate the chemical space autonomously toward optimal molecules using information from comparably few structures along the way, as in evolutionary AI inspired by the principles of natural selection and evolution. Whereas evolution-inspired methods aimed at design of drug candidates have been explored since the early 1990s, we have, over the last 15 years, developed the first method of this kind able to handle all kinds of molecules, ranging from organic drug-like molecules to transition metal catalysts. This is the sole de novo method experimentally verified for transition-metal compounds.
Highlights from the Development of Evolutionary AI for Molecular Design:
The first general-purpose de novo method reported (2012).
User-controlled synthetic accessibility of designed structures (2014).
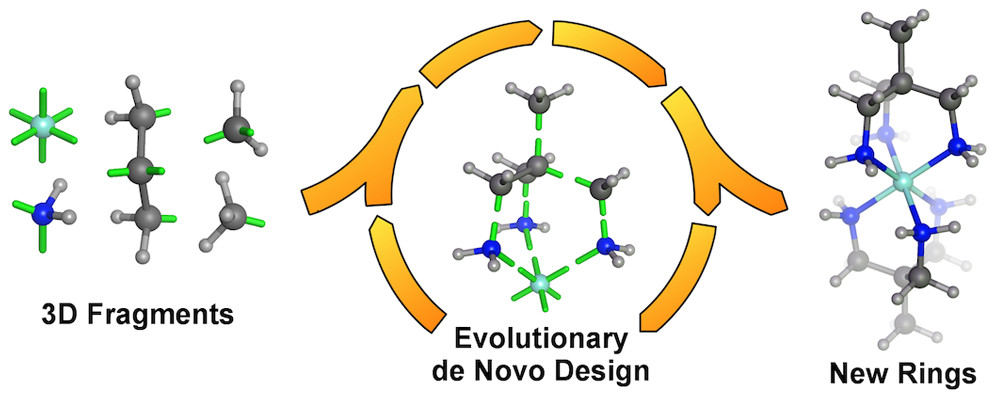
Automated construction of high-quality 3D molecular geometries (2014).
On-the-fly formation of cyclic structures, including metal chelates, during automated de novo design (2015).
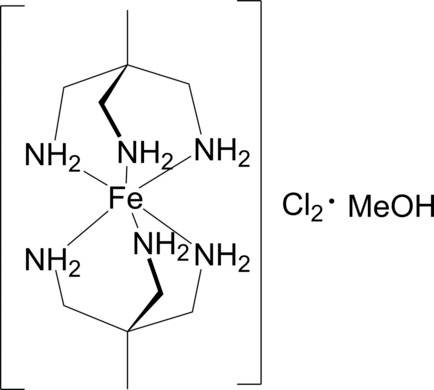
The first de novo designed inorganic molecule experimentally verified to reflect the intended property (designed: 2015, verified: 2018).
Open-source distribution of de novo design software package (2019).
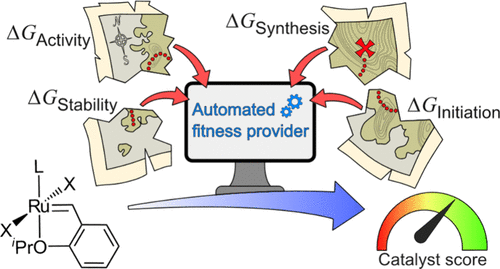
First principles-based fitness/scoring function for catalysis (2024).
Highlights from Ongoing Developments of Methods for Molecular Design and Discovery:
Combinations of exploitative and explorative, more creative, methods: Evolutionary and generative AI.
Multi-factor design criteria for catalysis.
ML-based surrogate models of target-inhibitor binding free energies.
Experimental follow-up of designed candidates using robotic, high-throughput experimentation at the HTE@UiB facility, located in our labs (see film).
We focus on design and development of catalysts and processes for sustainable chemical manufacturing from renewable resources. Examples of processes studied:
Production of alpha-olefins from fatty acid esters. Example: A Heterogeneous Catalyst for the Transformation of Fatty Acids to α‑Olefins. Chatterjee, A.; Jensen, V. R., ACS Catalysis 2017, 7, 7784-7789. DOI:10.1021/acscatal.6b03460.
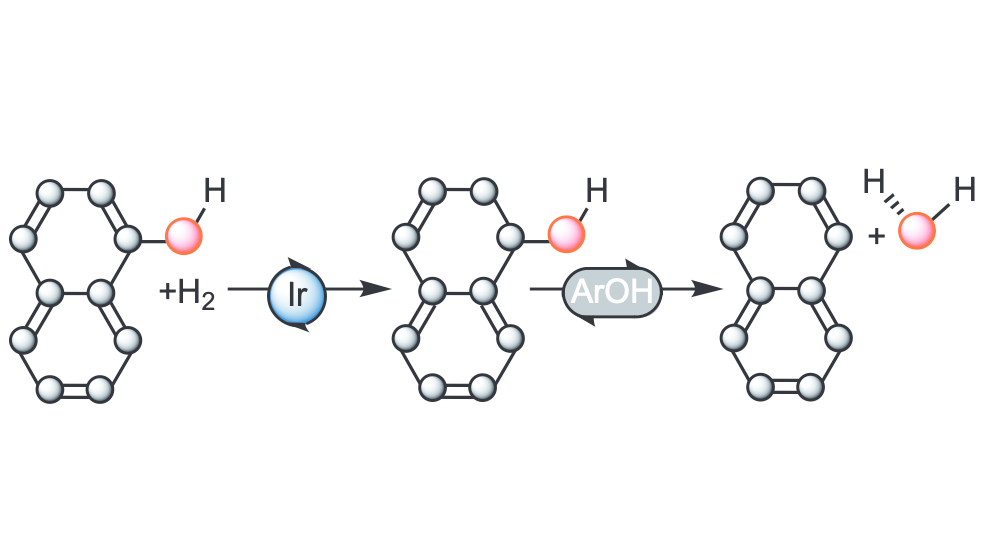
Aromatic hydrocarbons from lignin-derived phenolic compounds. Example: Molecular Insight into Hydrodeoxygenation of Naphthols: Iridium-Catalyzed Ring Hydrogenation and Substrate-Catalyzed Dehydration. Gjermestad, C. S.; Kusumoto, S.; Flores-Linares, I.; Occhipinti, G.; Le Roux, E.; Ando, H.; Nozaki, K.; Jensen, V. R.. Organometallics.2025, ASAP article. DOI:10.1021/acscatal.4c06212.
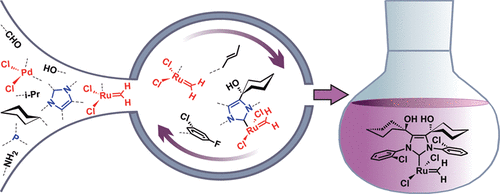
Olefin metathesis. Olefin metathesis is the most versatile method known for the construction and manipulation of carbon-based frameworks. Moreover, with an unmatched atom economy (little waste), a scope that spans across chemistry and chemical biology, and benign reaction conditions, olefin metathesis holds enormous potential in sustainable chemical manufacturing.
We have worked with olefin metathesis class for over 20 years, and organized, together with the group of Prof. Deryn E. Fogg (Universities of Ottawa and Bergen), the 2023 edition of the International Symposium on Olefin Metathesis and Related Chemistry, ISOM-XXIV, in Bergen. A cornerstone of our work has been obtaining and leveraging fundamental insights in the design and development of new catalysts.
A few highlights from our early work on olefin metathesis:
Landmark multivariate classification and QSAR/QSPR relationships for ruthenium-based catalysts. Quantitative Structure—Activity Relationships of Ruthenium Catalysts for Olefin Metathesis. Occhipinti, G.; Bjørsvik, H. R.; Jensen, V. R. J. Am. Chem. Soc.2006, 128, 6952-6964. DOI: 10.1021/ja060832i
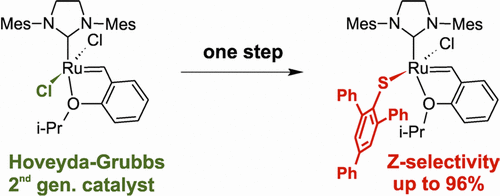
A whole new family of computationally designed stereoselective catalyst. Example: Simple and Highly Z-Selective Ruthenium-Based Olefin Metathesis Catalyst. Occhipinti, G.; Hansen, F. R.; Törnroos, K. W.; Jensen, V. R. J. Am. Chem. Soc. 2013, 135, 3331-3334. DOI: 10.1021/ja311505v
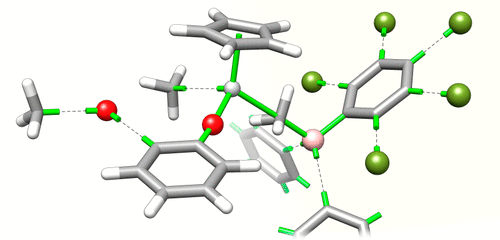
Over the last few years, we have focused on improving the stability of the olefin metathesis catalysts. Despite its potential, olefin metathesis is notorious for the severe problems arising from catalyst decomposition, which restrict both productivity and selectivity, and indeed industrial uptake. Catalysts resistant to decomposition would eliminate the chief obstacle to implementation of metathesis in the industrial context, with major implications for utilization of renewable feedstocks, and production of specialty chemicals and active pharmaceutical ingredients. To help realize this potential we have, together with the group of Prof. Deryn E. Fogg (Universities of Ottawa and Bergen), conducted a campaign to uncover the detailed mechanisms, controlling factors, and kinetic role of two intrinsic decomposition pathways (that is, pathways that involve no species beyond those within the catalytic cycle itself).
A few highlights from the campaign:
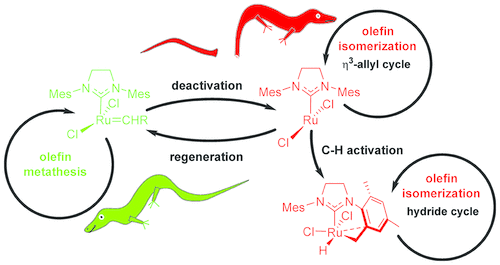
Connection between catalyst decomposition and regeneration. Example: Loss and Reformation of Ruthenium Alkylidene: Connecting Olefin Metathesis, Catalyst Deactivation, Regeneration, and Isomerization. Engel, J.; Smit, W.; Foscato, M.; Occhipinti, G.; Törnroos, K. W.; Jensen, V. R. J. Am. Chem. Soc.2017, 139, 16609-16619; DOI: 10.1021/jacs.7b07694.
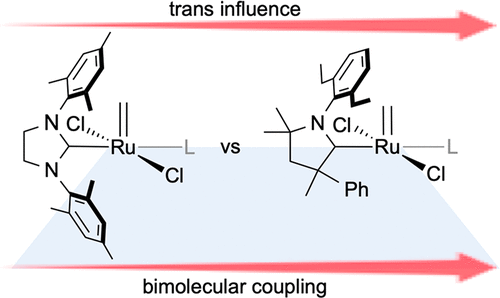
Individual decomposition reactions and mechanisms. Bimolecular Coupling in Olefin Metathesis: Correlating Structure and Decomposition for Leading and Emerging Ruthenium-Carbene Catalysts. Nascimento, D. L.; Foscato, M.; Occhipinti, G.; Jensen, V. R.; Fogg, D. E.J. Am. Chem. Soc.2021, 143, 11072-11079. DOI: 10.1021/jacs.1c04424.
Bringing the pieces together. Example: The Janus face of high trans-effect carbenes in olefin metathesis: gateway to both productivity and decomposition. Occhipinti, G.; Nascimento, D. L.; Foscato, M.; Fogg, D. E.; Jensen, V. R.Chem. Sci.2022, 13, 5107-5117. DOI: 10.1039/d2sc00855f.